Main Body
Miscellaneous Glomerular Diseases
In addition to the common causes of proteinuric kidney disease in dogs, there are other patterns of glomerular injury that are rare or insufficiently characterized. With time, we hope to develop standardized criteria for diagnosis of these diseases and better understand their pathogeneses. Some of these diseases still have clinical features based on what we have gleaned from our database of cases, whereas the rarity of other diseases have precluded our ability to report the relevant clinicopathologic data.
MESANGIOPROLIFERATIVE GLOMERULONEPHRITIS WITH IMMUNE COMPLEXES
- Glomerular disease characterized by frequent mesangial hypercellularity (>3 nuclei) and associated mesangial matrix expansion secondary to the presence of immune complex deposits that are themselves limited to mesangial zones. Diagnosis requires TEM and IF to verify the presence of the immune complexes.
- There is often segmental sclerosis observed on histology.
Cases with mesangial expansion and / or hypercellularity without identifiable immune complex deposits should be given a descriptive diagnosis of “Glomerulopathy characterized by mesangial cell proliferation.”
Clinical Features
- Initial sign is typically moderate to marked proteinuria (UPC > 2), although median UPC is lower than most other categories of glomerular disease, and rare cases can have a UPC as low as 0.5. Dogs with mesangioproliferative GN are unlikely to have a UPC higher than ~15, and they are more likely to have normal serum albumin concentration compared with many of the other categories of glomerular disease.
- Dogs with mesangioproliferative GN are more likely to be non-azotemic at the time of biopsy than other categories of glomerular disease.
- While rare cases have been identified in dogs as young as 4 months of age, dogs <1 year of age are unlikely to have mesangioproliferative GN.
- No breed predisposition is currently recognized.
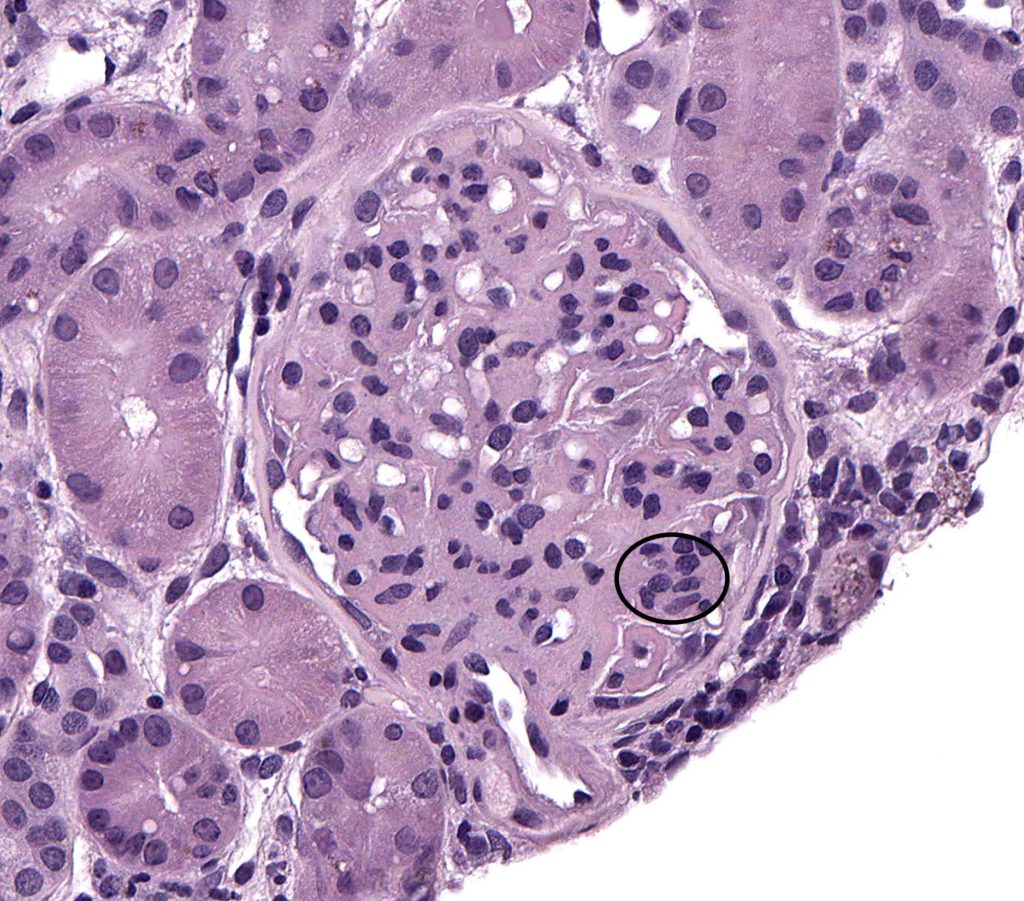
FIG.1A (HE): Moderate mesangial cell hypercellularity (greater than 3 nuclei in close apposition) within an expanded mesangial matrix (circled).
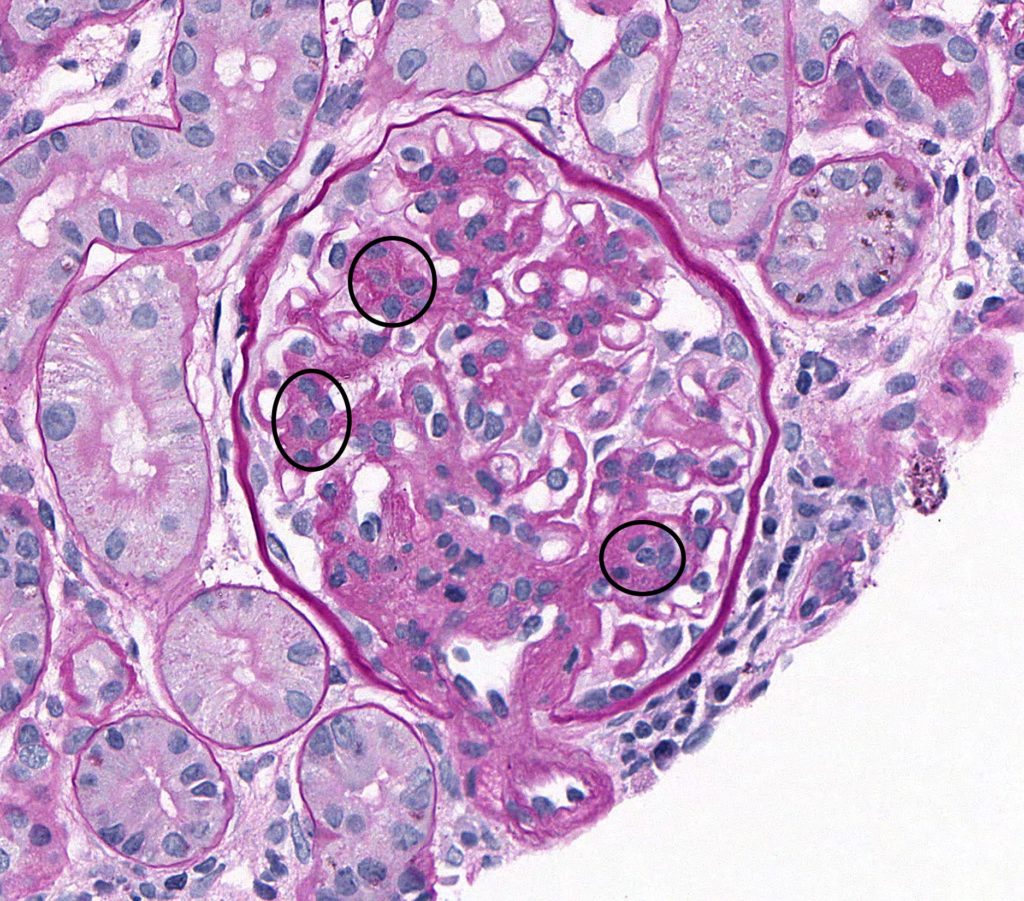
FIG.1B (PAS): Increased numbers of mesangial cells within an expanded matrix (circled).
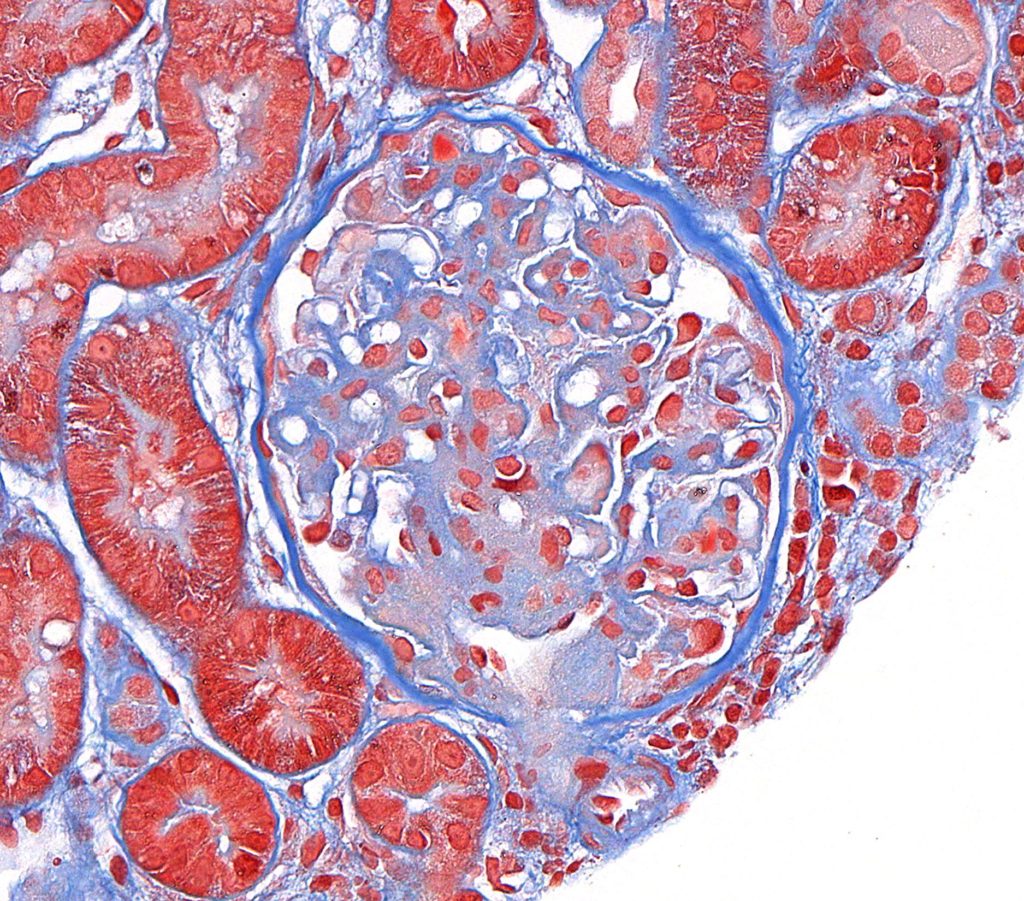
FIG.1C (MT): Mild to moderate mesangial cell hyperplasia with matrix expansion.
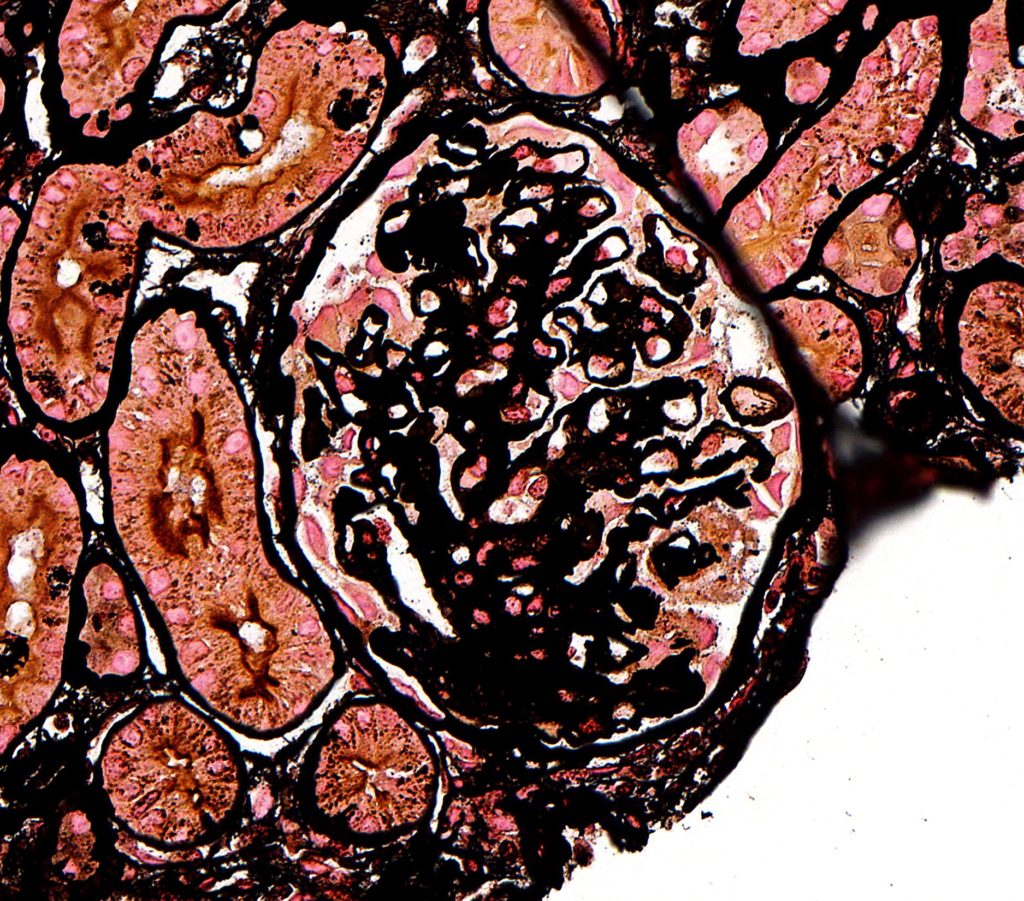
FIG.1D (JMS): The GBM is irregularly thickened. Please note that this JMS stain is dark due to prolonged time in silver.
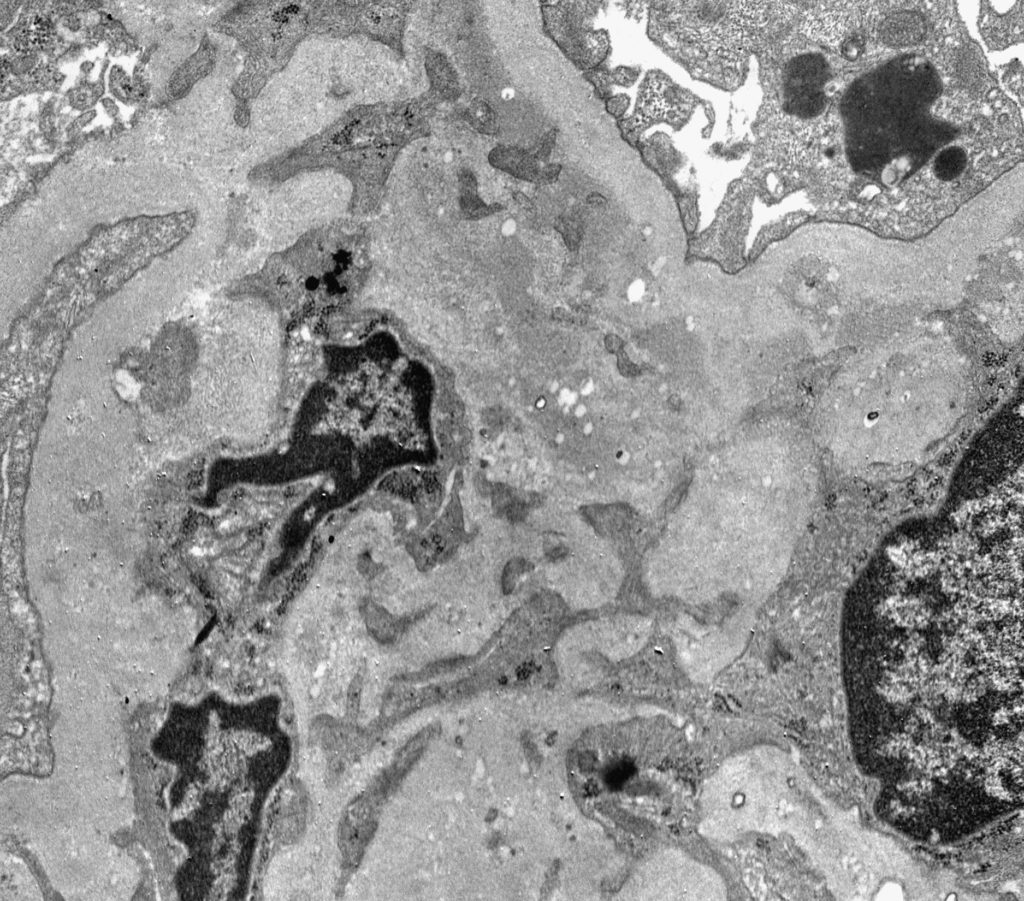
FIG.1E (TEM): Portions of 3 mesangial cell nuclei in close appostion (mesangial hyperplasia). Note multiple electron dense deposits withih the mesangial area, often just beneath the GBM (paramesangial).
FIG.1F (TEM): Colorized version of above TEM image with electron dense deposits (black) between the mesangial matrix (blue) and the paramesangial GBM (green). The podocytes are pink.
MINIMAL CHANGE DISEASE (Minimal change glomerulopathy)
- Minimal change disease is an uncommon (in dogs), acquired, potentially reversible, podocytopathy. May be drug-induced (reported in dogs given the tyrosine kinase inhibitor masitinib), idiopathic, or presumed immune-mediated (steroid responsive MCD in children).
- Characterized by relatively normal (hence the name) glomeruli via light microscopy and absence of specific tubulointerstitial or vascular lesions. The detection of glomerular changes and final diagnosis require TEM that will show global podocyte foot process effacement.
- Affected dogs are markedly proteinuric. Azotemia has been rarely reported due to presumed associated acute tubular necrosis.
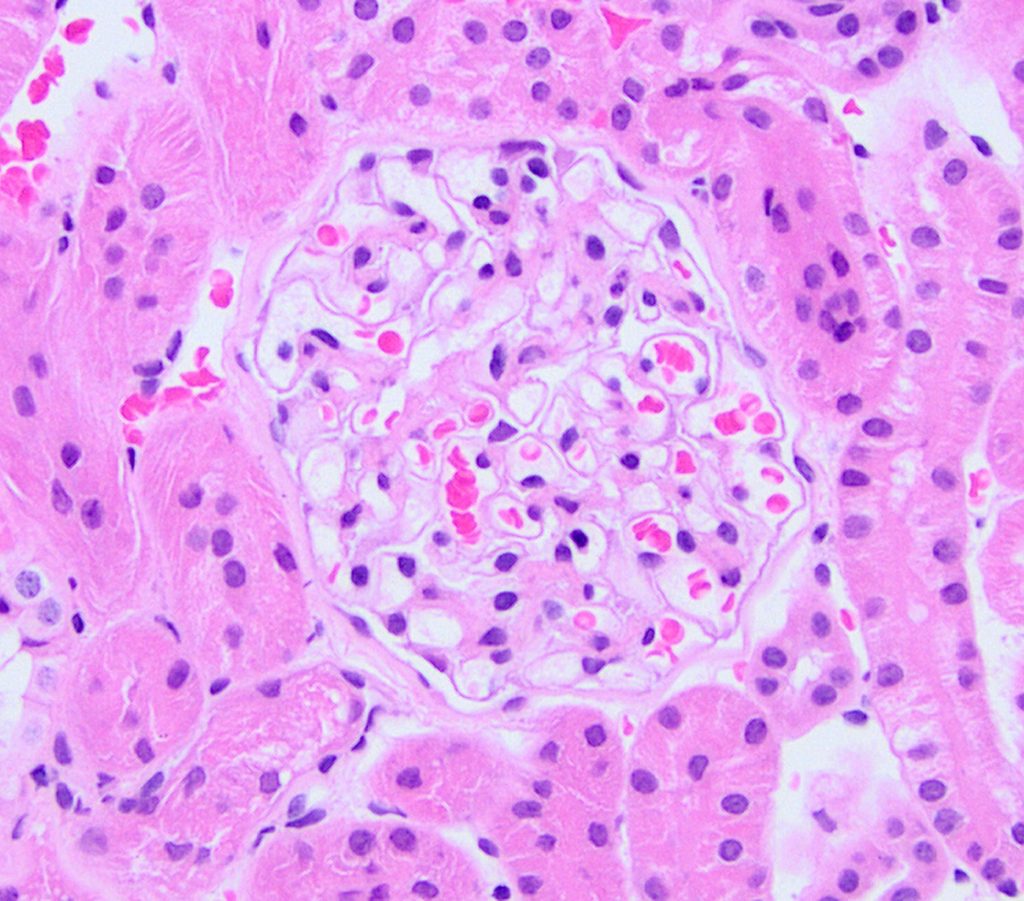
FIG.2A (HE): The glomerulus is normocellular.
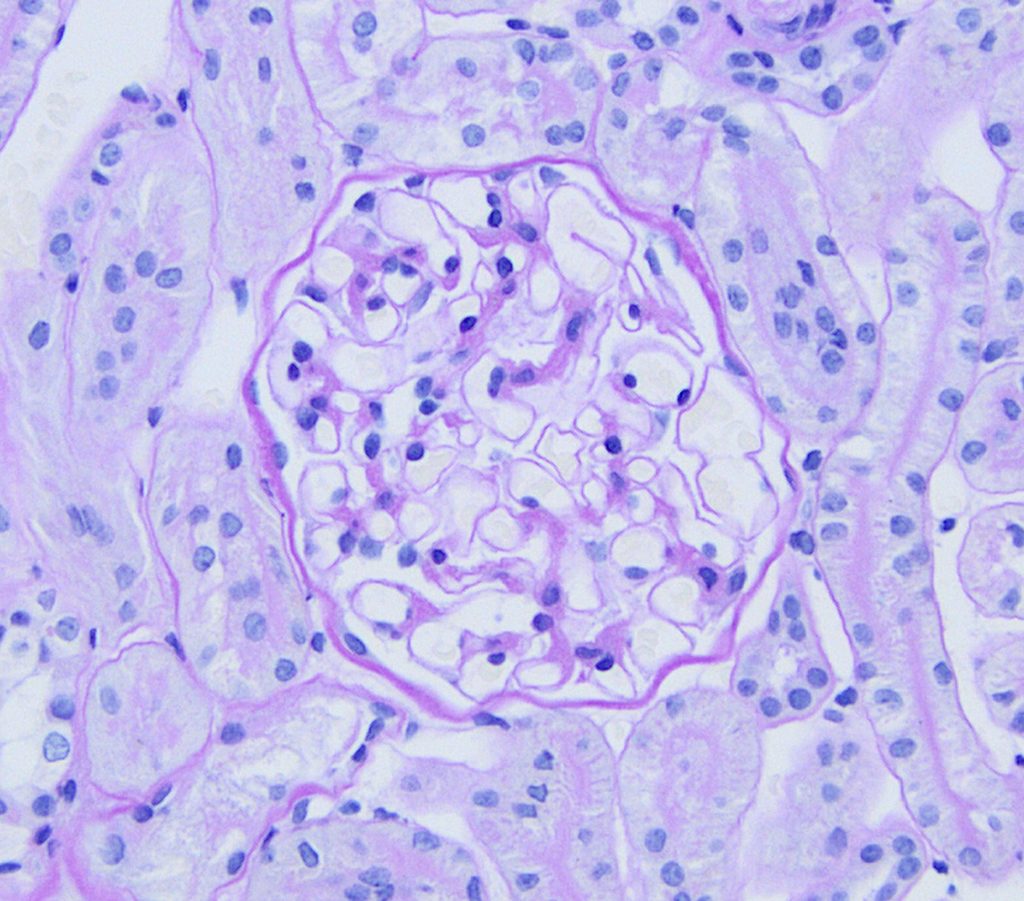
FIG.2B (PAS): The glomerulus is normocellular and the mesangium and GBM are unremarkable.
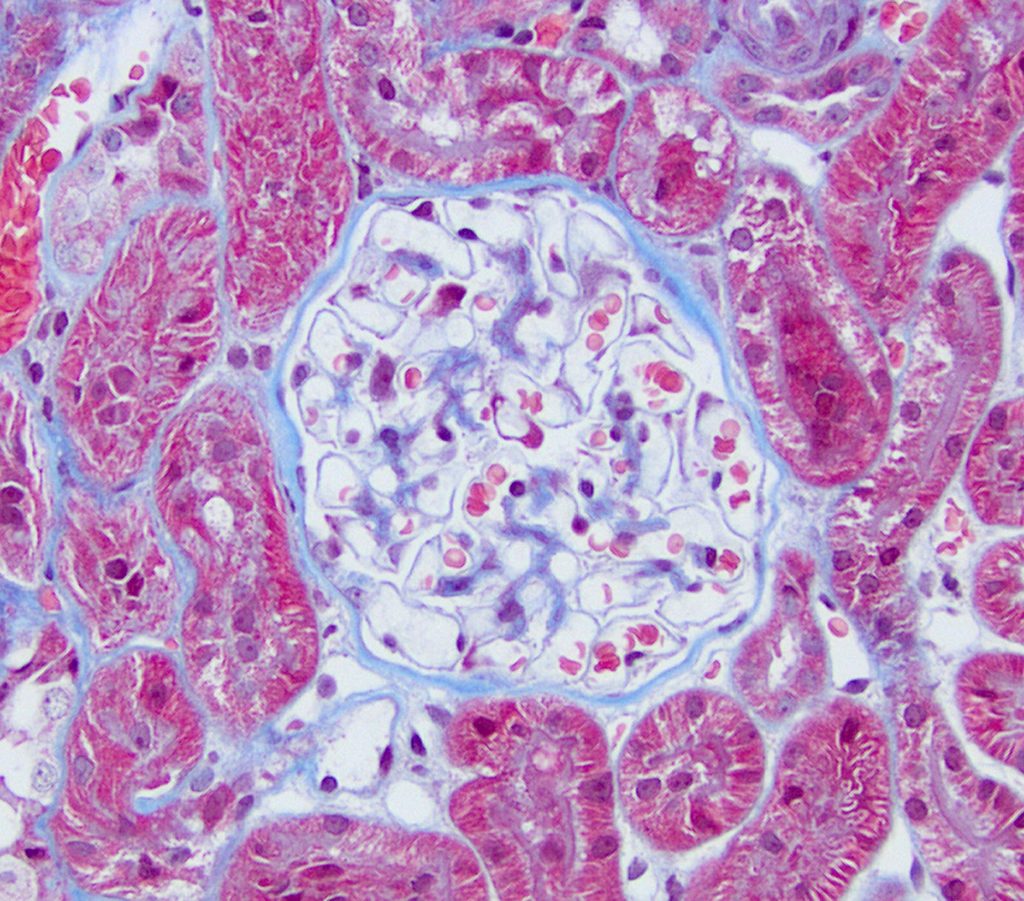
FIG.2C (MT): The glomerulus is histologically normal. Fuschinophilic (red) deposits are not osberved. The GBM is thin and uniform, the mesangium is normal, and there is no fibrosis in the surrounding interstitium.
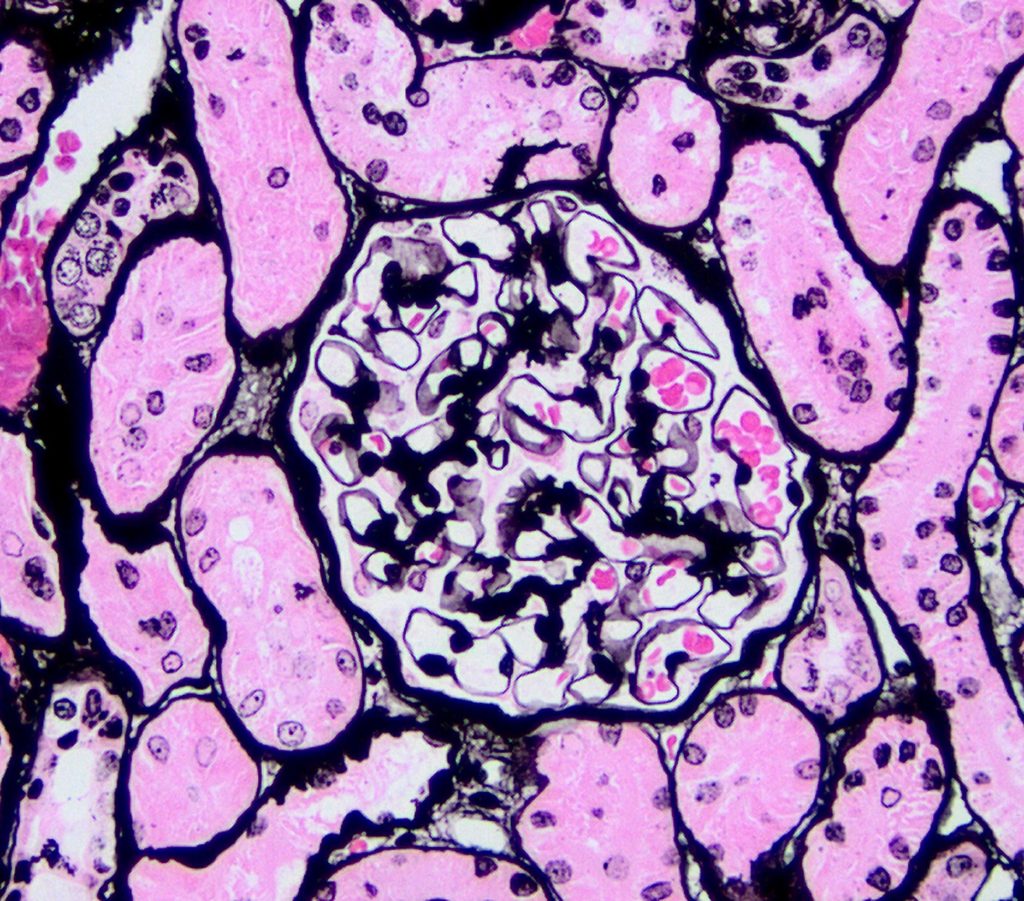
FIG.2D (JMS): Capillary walls are of normal thickness; there is no evidence of GBM remodeling.
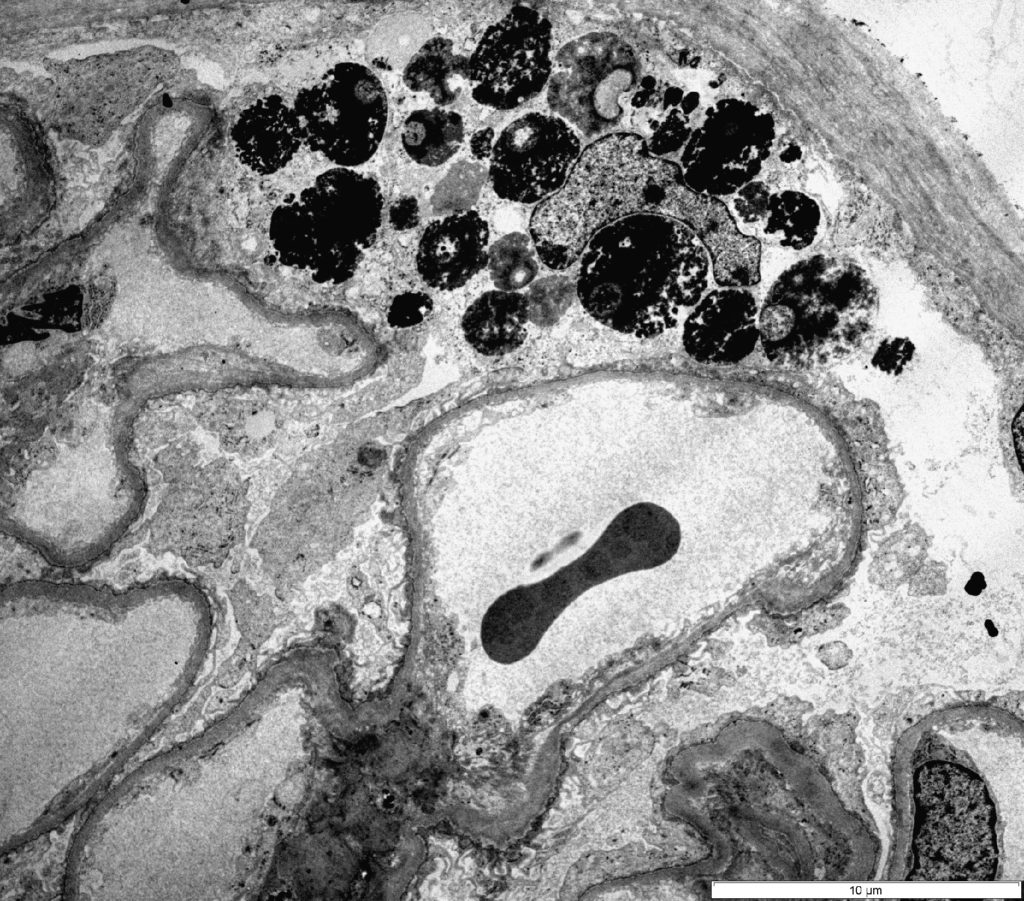
FIG.2E (TEM): Ultrastructural evidence of diffuse and global podocyte injury is present. In this case, one podocyte contains large electron dense lysosomes, and although difficult to appreciate at this low magnification, all podocytes exhibit foot process effacement.
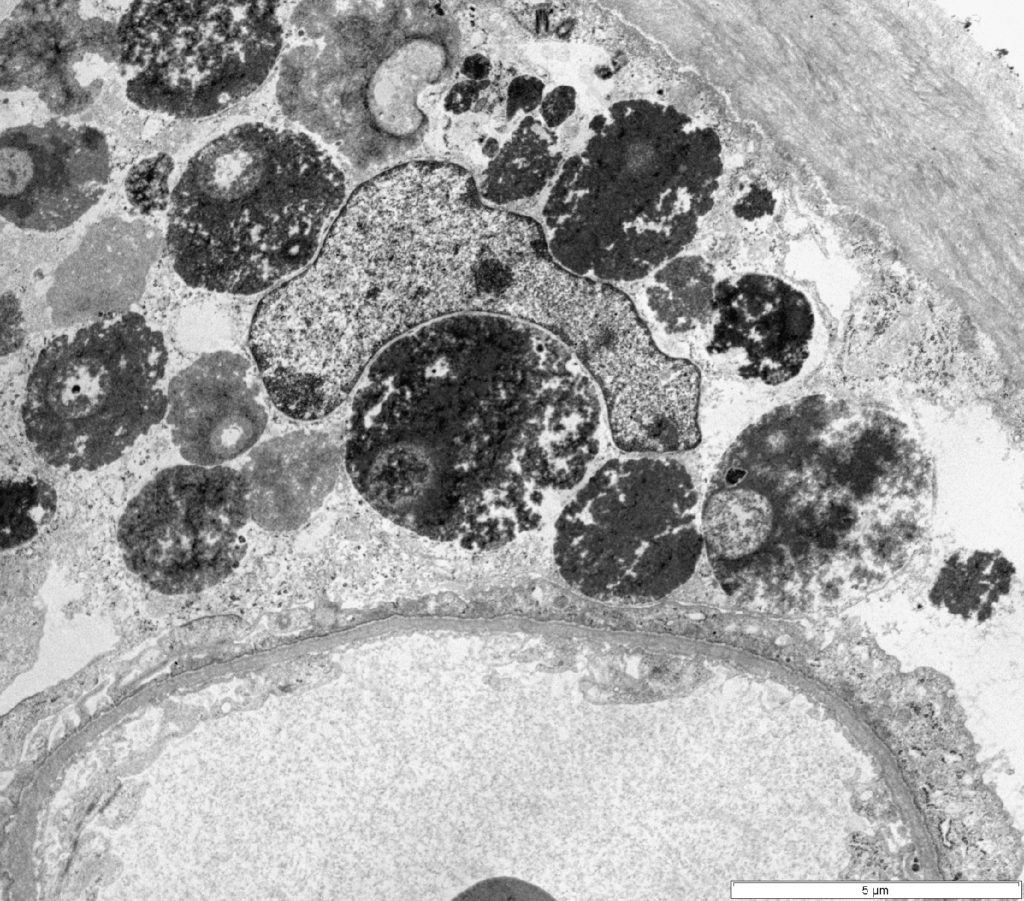
FIG.2F (TEM): Severe podocyte injury with foot process effacement, protein resorption droplets, and cell lysis. The endothelium is unremarkable, and neither electron dense deposits nor fibrils are evident.
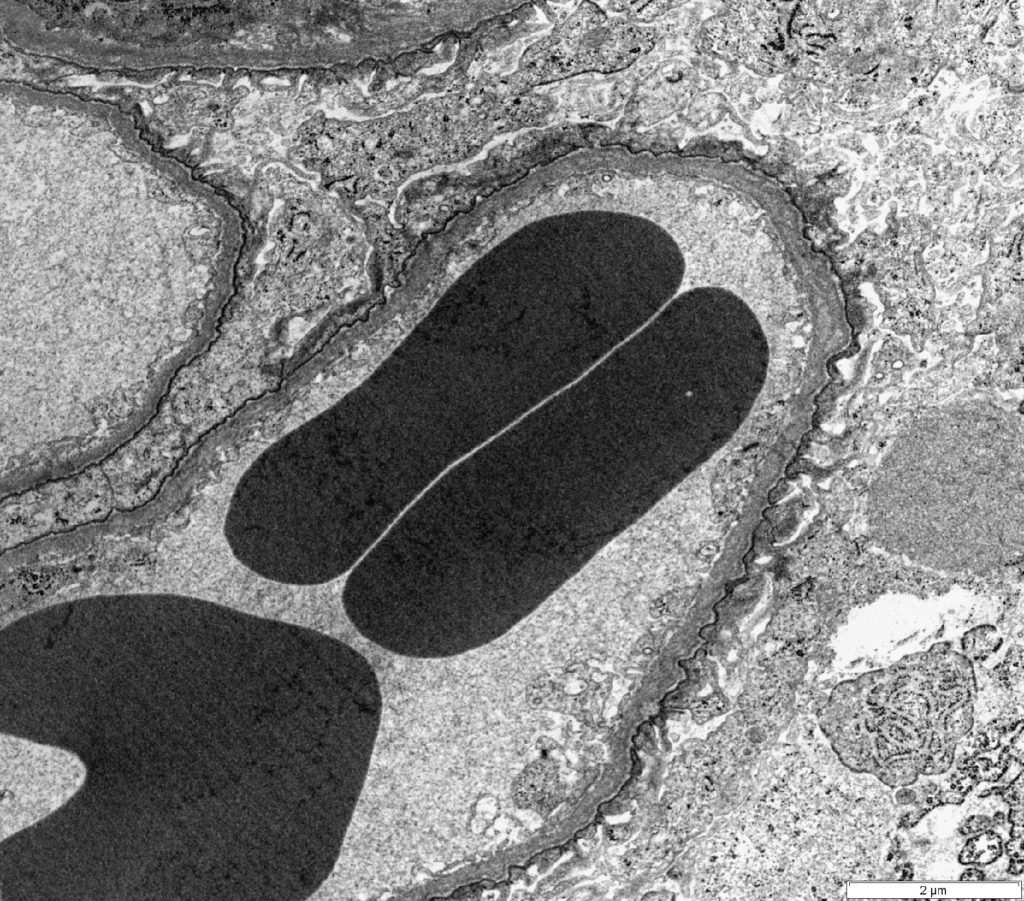
FIG.2G (TEM): There is diffuse foot process effacement. Podocytes are hypertrophied, with podocyte cytoplasm filling Bowman’s space.
GLOMERULAR LIPIDOSIS
- Presence of large “foamy” cells found in one or multiple lobules of the glomerular tufts. The foamy appearance is given by the presence of intracytoplasmic vacuole that are sudanophilic (lipid material) but are clear with other routine staining (HE, PAS, MT and JMS).
- The origin of the cells is unknown but mesangial and endothelial origin has been proposed.
- Historically thought to be an incidental finding not affecting the glomerular function. A more severe disease is presumed if the lesion is detected in many glomeruli and if the glomerular architecture is severely effaced.
- Sometimes detected as sole lesion in proteinuric dogs.
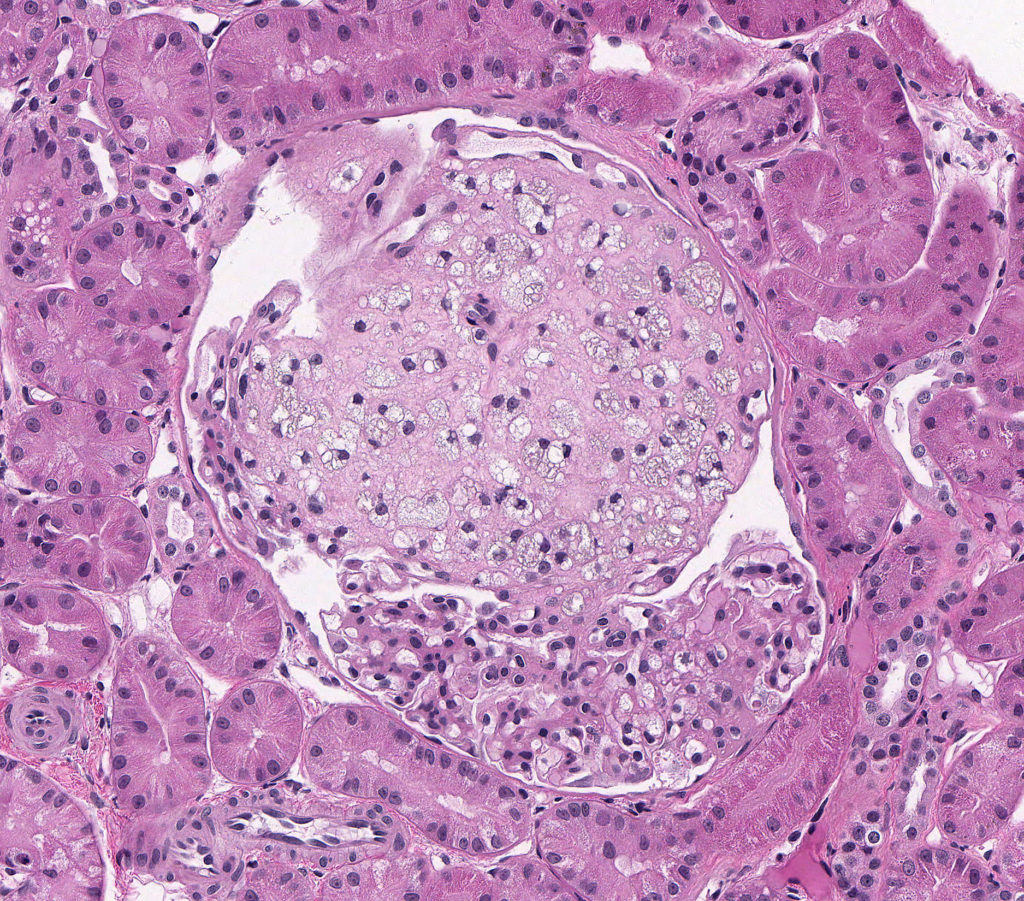
FIG.3A (HE): The glomerular tuft architecture is segmentally distorted and effaced with large “foamy” cells.
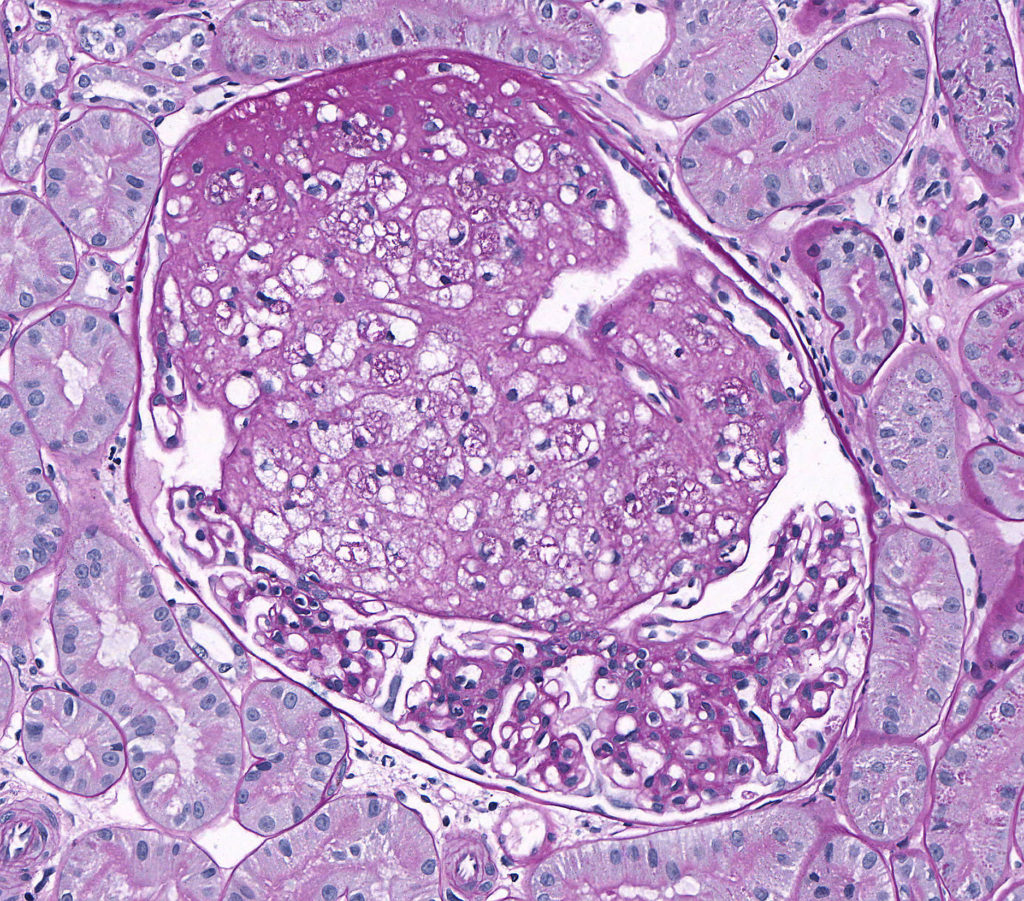
FIG.3B (PAS): The glomerular tuft architecture is segmentally distorted and effaced with large “foamy” cells. the remainder of the tuft appears fairly normal but compressed.
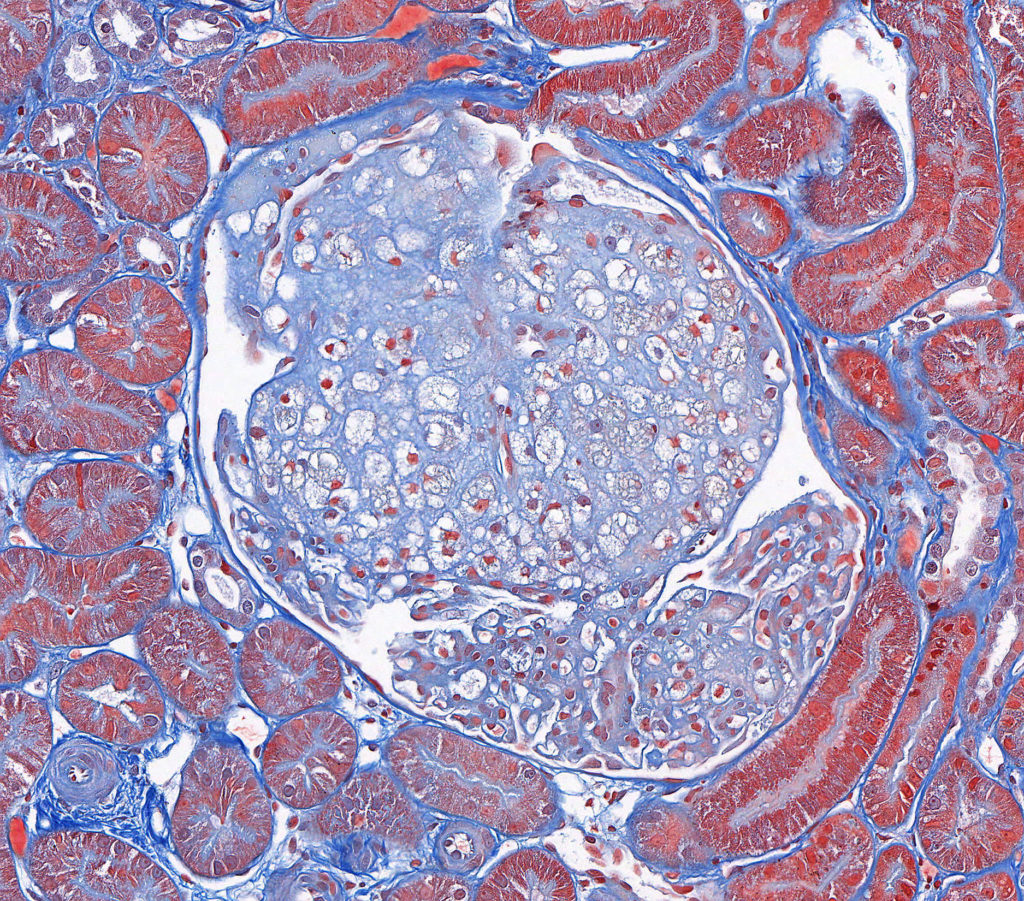
FIG.3C (MT): The glomerular tuft architecture is segmentally distorted and effaced with large “foamy” cells.
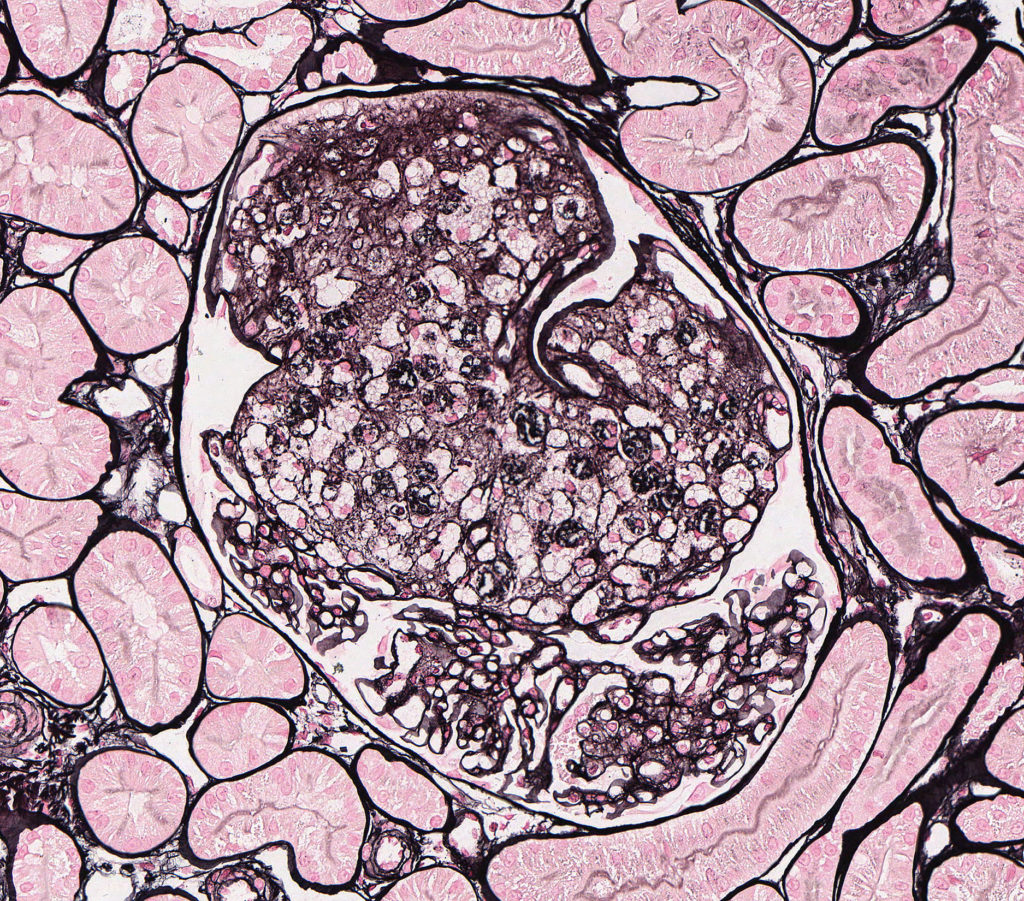
FIG.3D (JMS): The glomerular tuft architecture is segmentally distorted and effaced with large “foamy” cells. The capillary walls of the remaining portion of the glomerulus are of normal thickness and have a smooth contour.
THROMBOTIC MICROANGIOPATHY
- Lesions of renal thrombotic microangiopathy (TMA) are due to endothelial injury of glomerular capillaries and arterioles, which can manifest clinically as acute kidney injury with microangiopathic hemolytic anemia and thrombocytopenia. Microangiopathic hemolytic anemia is a condition in which erythrocytes are damaged and destroyed when traveling through small vessels. This damage can be due to small intravascular fibrin thrombi (as can occur in Disseminated Intravascular Coagulation) or due to severe endothelial damage from toxin exposure (e.g. envenomation), infection, immune-mediated (both complement system and antibody mediated) and severe acute hypertension, among other causes. Renal TMA is rare and is characterized primarily by injury to glomerular capillaries and afferent arterioles with occasional inclusion of larger arteries. Canine diseases that include this lesion are few but can be seen in cases of severe hypertension, hemolytic-uremic syndrome, and most notably in idiopathic cutaneous and renal glomerular vasculopathy (CRGV). CRGV has been documented in racing greyhounds in the US and a variety of other breeds in the UK. Affected dogs present with cutaneous ulceration of the distal limbs, thrombocytopenia and acute kidney injury. Additionally, because of the association between TMA and acute hypertension, TMA lesions can be superimposed on other types of renal diseases such as immune complex mediated membranoproliferative glomerulonephritis. In these scenarios, both diseases should be diagnosed (for example: immune complex mediated MPGN with superimposed TMA) because the clinician should be aware that they are dealing with 2 types of processes.
- Light microscopic findings in cases of canine renal TMA might include:
- Endothelial swelling with narrowing of capillary lumens.
- Intra-capillary thrombi.
- Fragmented erythrocytes (schistocytes)
- Hyalinization and fibrinoid necrosis of afferent and intralobular arterioles and arteries.
- With progression there may be duplication of the glomerular basement membrane seen as double contours with silver stain. IF studies are consistent with non-specific staining.
- Ultrastructural findings might include:
- Endothelial swelling.
- Detachment from the underlying basement membrane and necrosis.
- Capillary lumens might contain platelets, fibrin, and cellular fragments.
- In acute TMA, there is extensive subendothelial lucency of the glomerular basement membrane. Although this latter lesion has been documented in humans and dogs, it is rarely seen because the acute stages are often missed.
- Chronically, there might be mesangiolysis and mesangial cell interpositioning with basement membrane duplication.
- In scenarios where TMA is the only disease process, electron dense deposits consistent with immune complexes are not present; however, if TMA is secondary to ICGN and hypertension, it is possible to observe electron dense deposits as well as TMA lesions in the EM specimen.
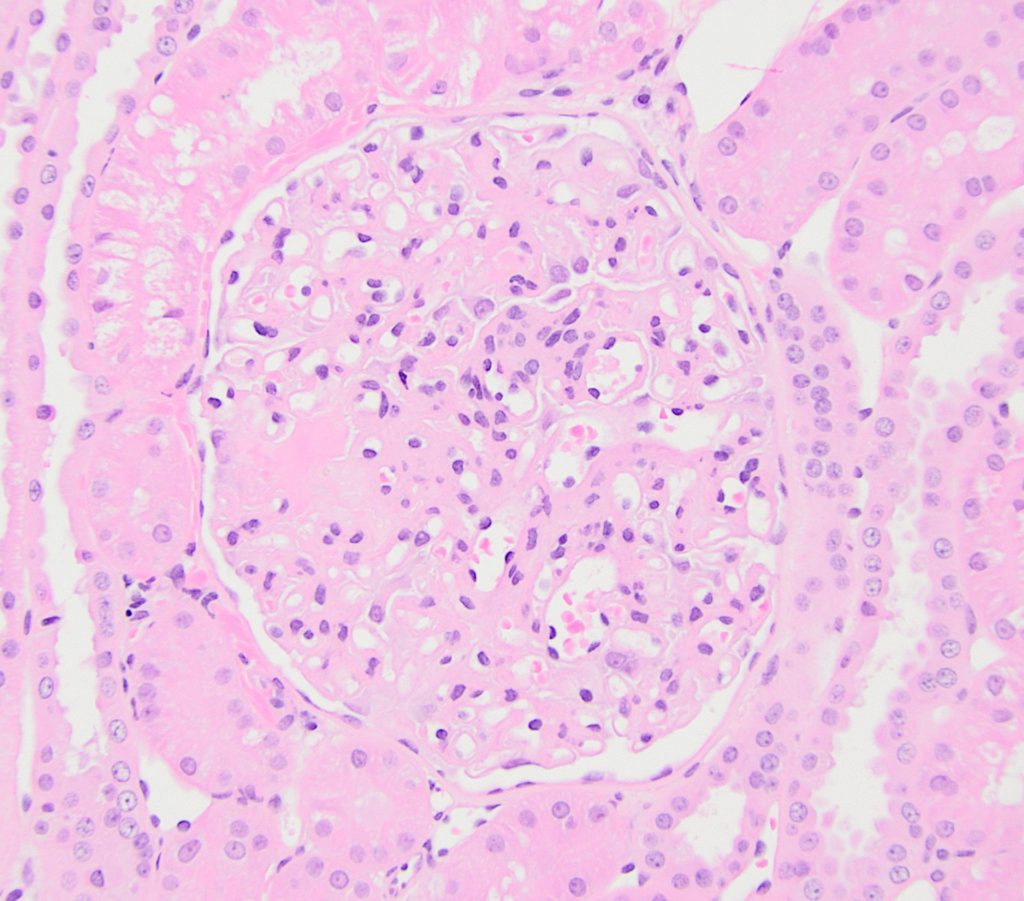
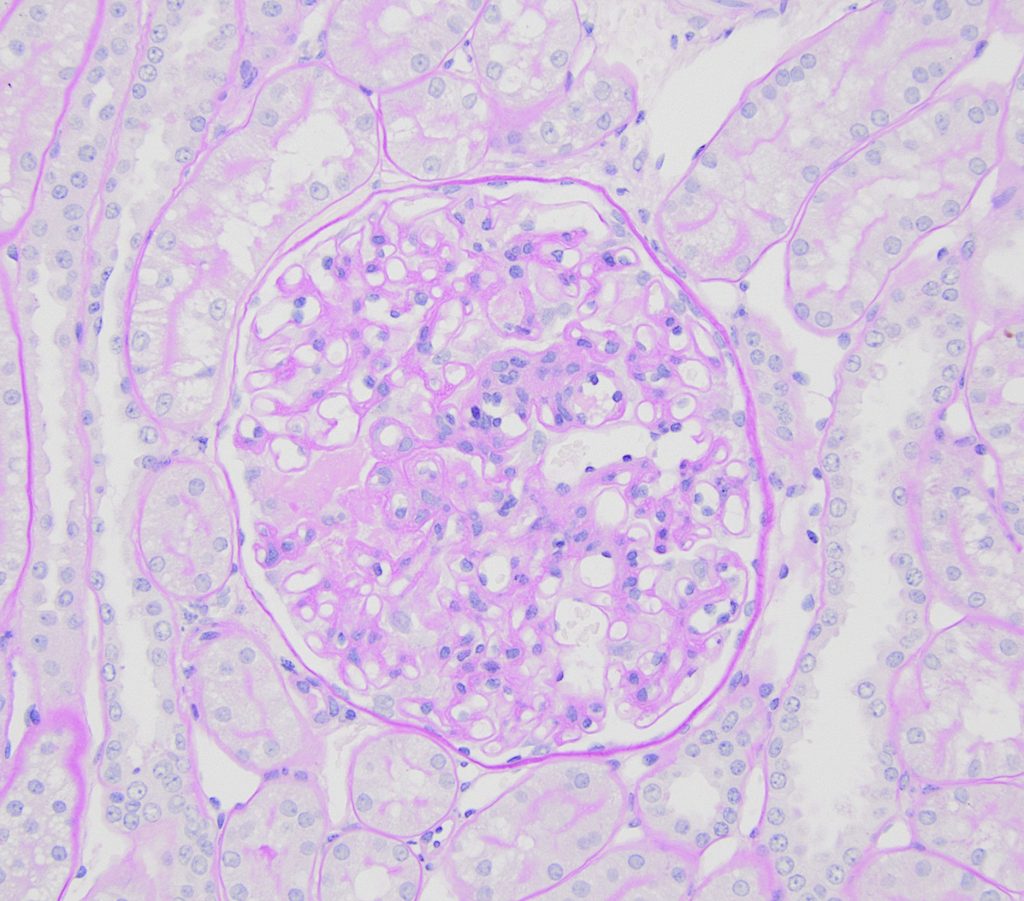
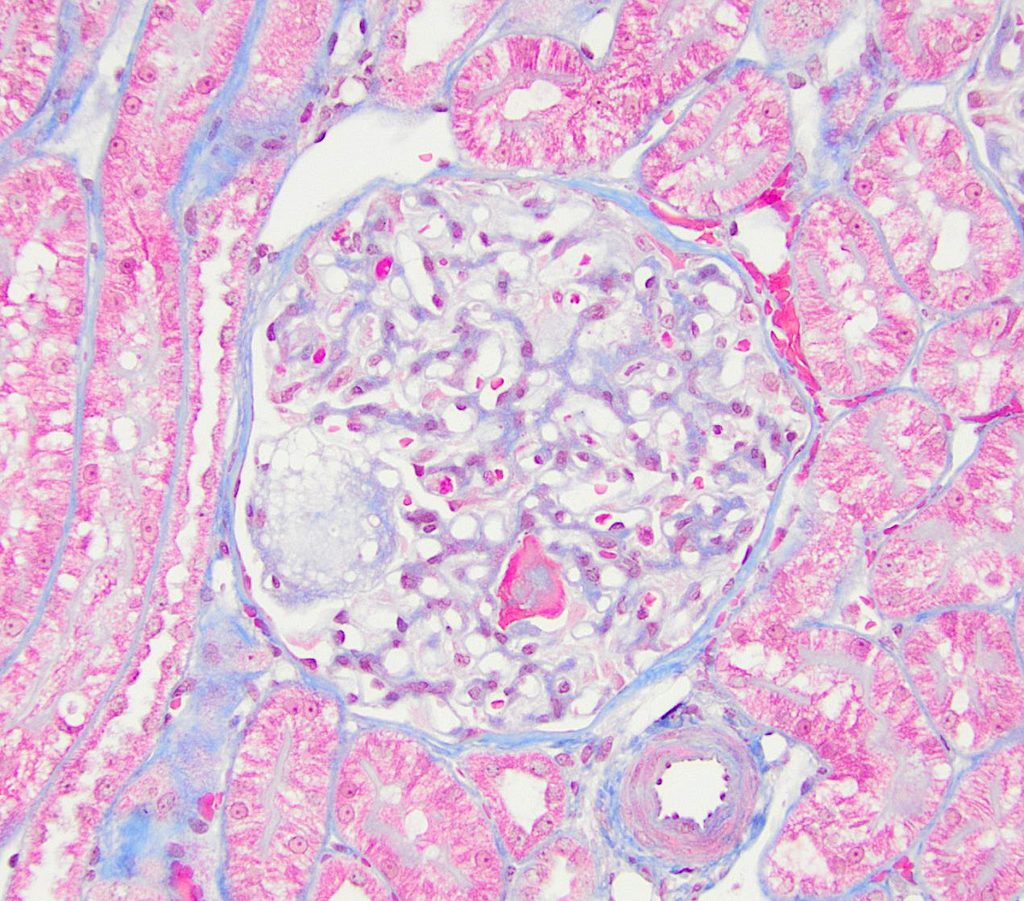
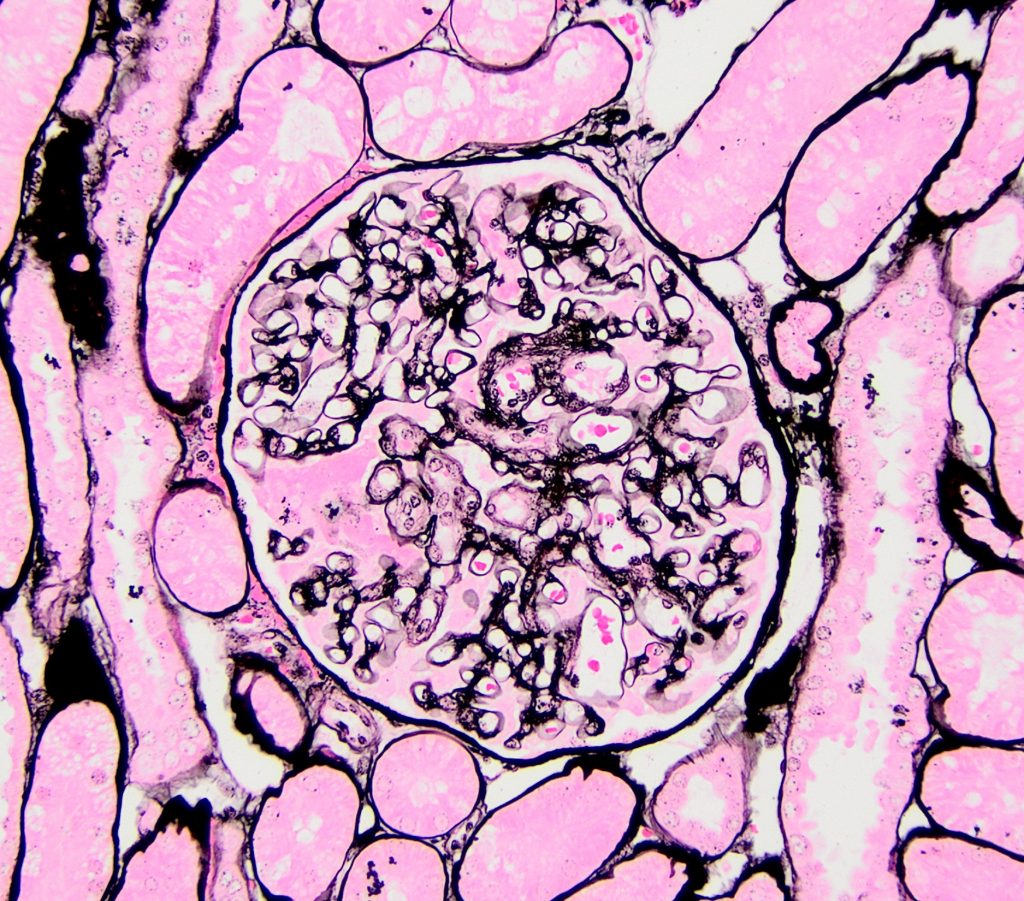
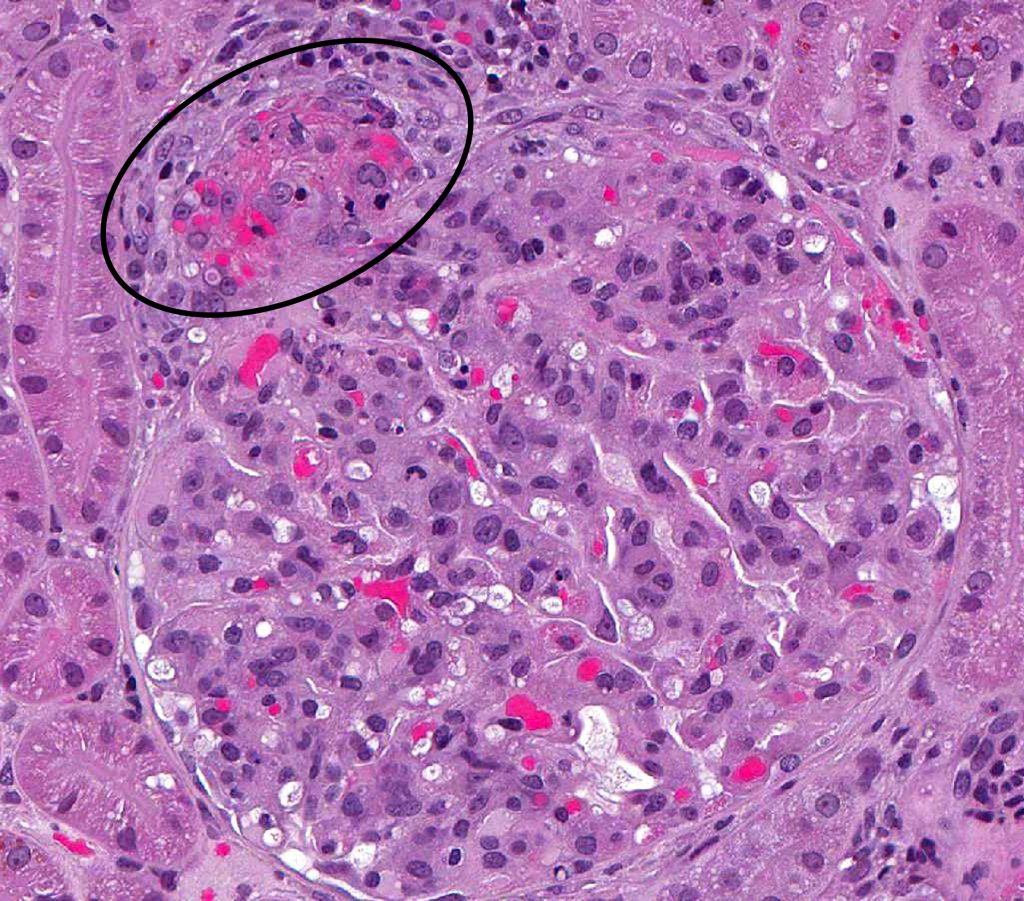
FIG.5A (HE): Thrombotic microangiopathic lesions can occur in patients that have underlying disease which might result in hypertension. In these scenarios, the vascular pole is usually affected (circled). 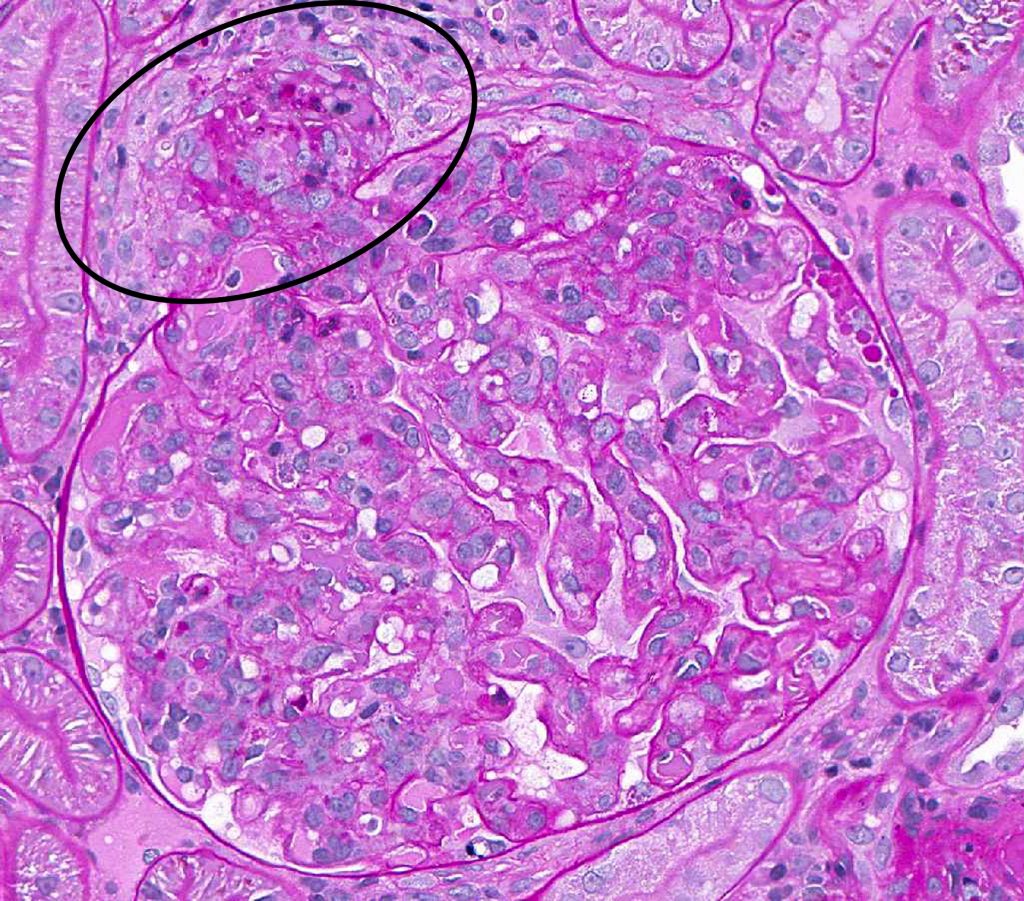 FIG.5B (PAS): In this stain, the fibrinoid degeneration of the arteriole is easily seen as fuschinophilic material in the wall (circled).
FIG.5B (PAS): In this stain, the fibrinoid degeneration of the arteriole is easily seen as fuschinophilic material in the wall (circled).
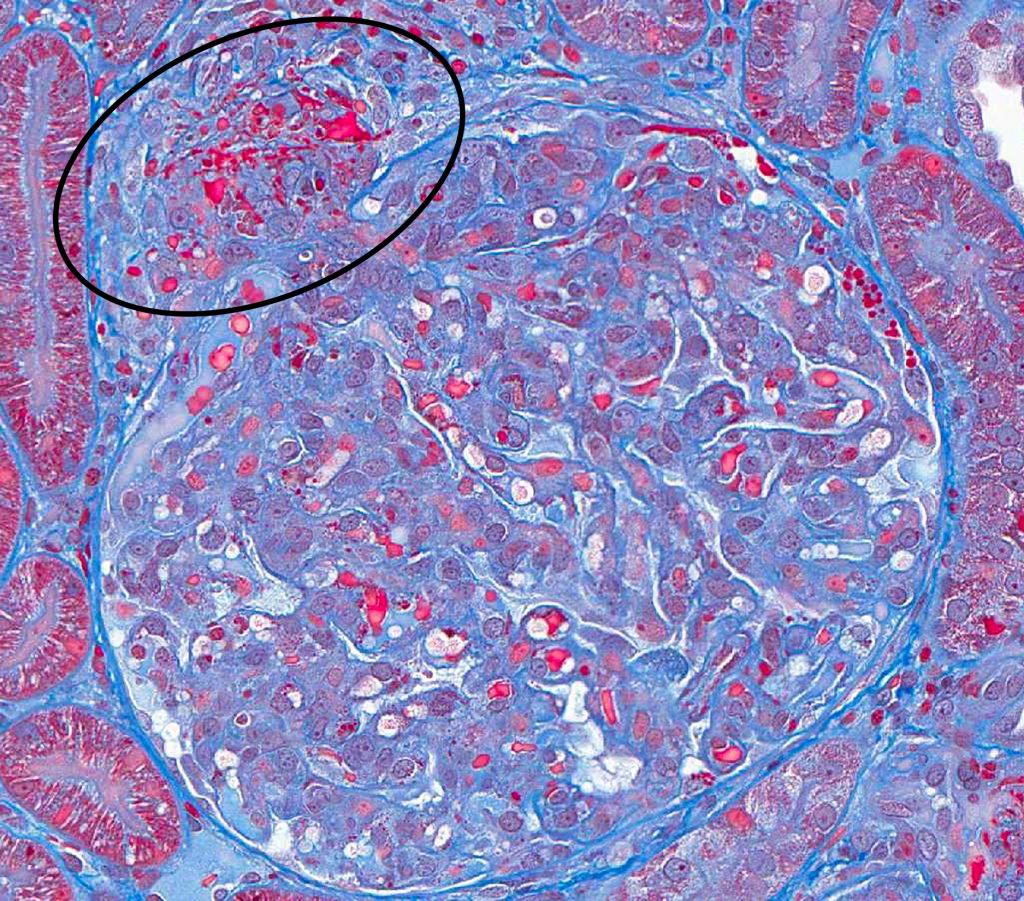
FIG.5C (MT): In this stain, the fibrinoid degeneration of the arteriole is easily seen as red material in the wall (circled). This stain also enable visualization of the fragmented red cells in the wall.
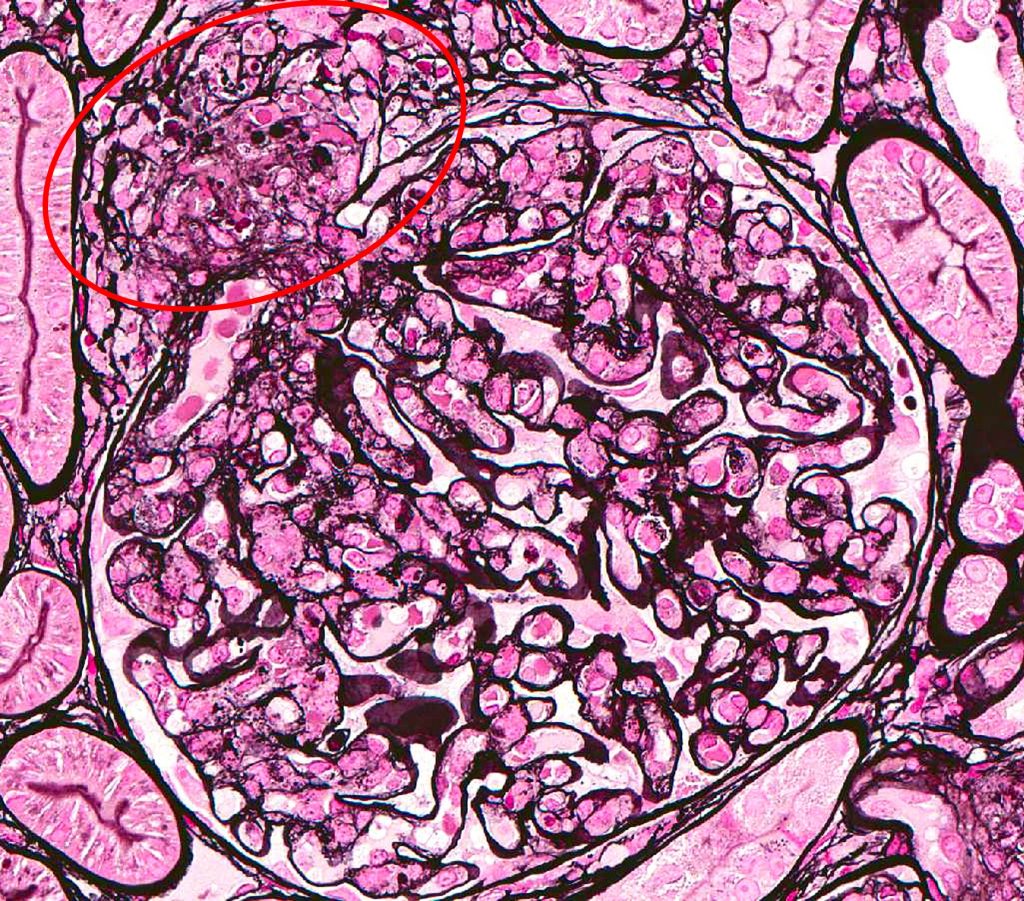
FIG.5D (JMS): In this stain, the fibrinoid degeneration of the arteriole is easily seen (circled).![]() FIG.5E (HE): Higher magnification of FIG.5A. Erythrocytes have been pushed into the wall of the afferent arteriole (black circle) and there is fibrinoid degeneration of the artertiolar wall (red circle). Pyknotic nuclear debris is also present in the wall (arrow).
FIG.5E (HE): Higher magnification of FIG.5A. Erythrocytes have been pushed into the wall of the afferent arteriole (black circle) and there is fibrinoid degeneration of the artertiolar wall (red circle). Pyknotic nuclear debris is also present in the wall (arrow).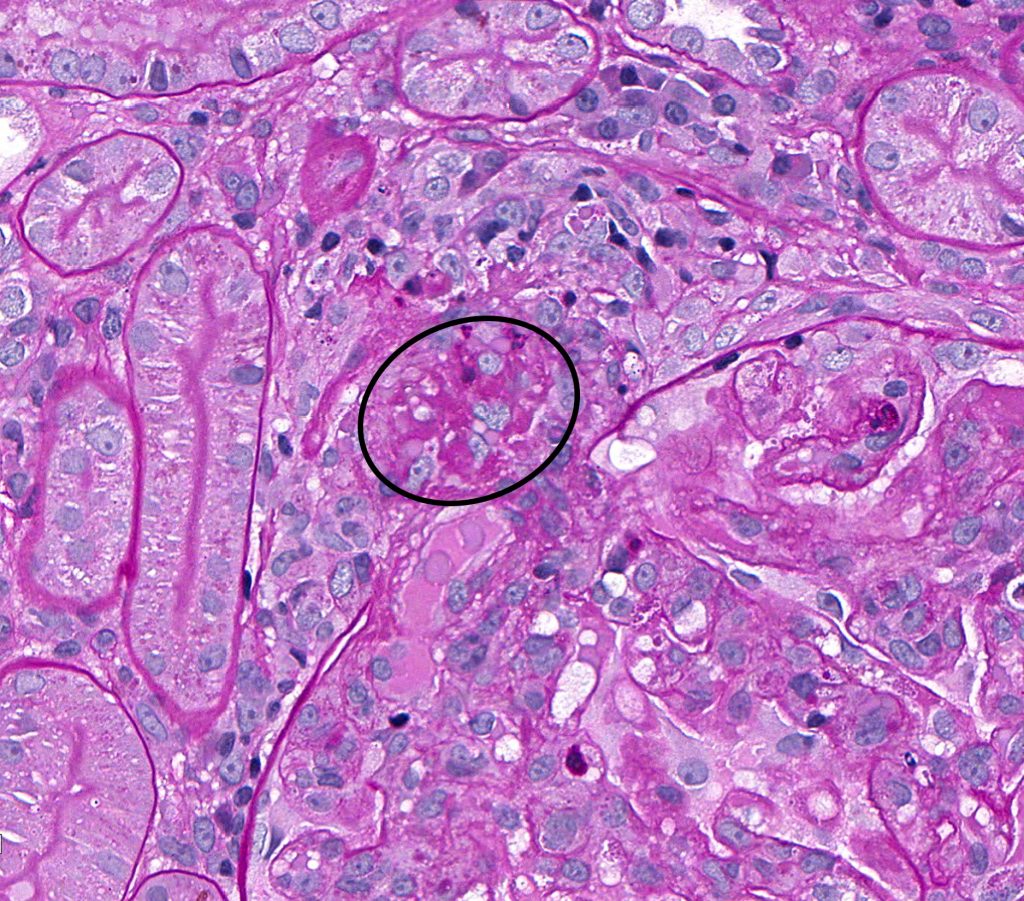 FIG.5F (PAS): Higher magnification of FIG.5B. In this stain, the fibrinoid degeneration of the arteriole is easily seen as fuschinophilic material in the wall (circled).
FIG.5F (PAS): Higher magnification of FIG.5B. In this stain, the fibrinoid degeneration of the arteriole is easily seen as fuschinophilic material in the wall (circled).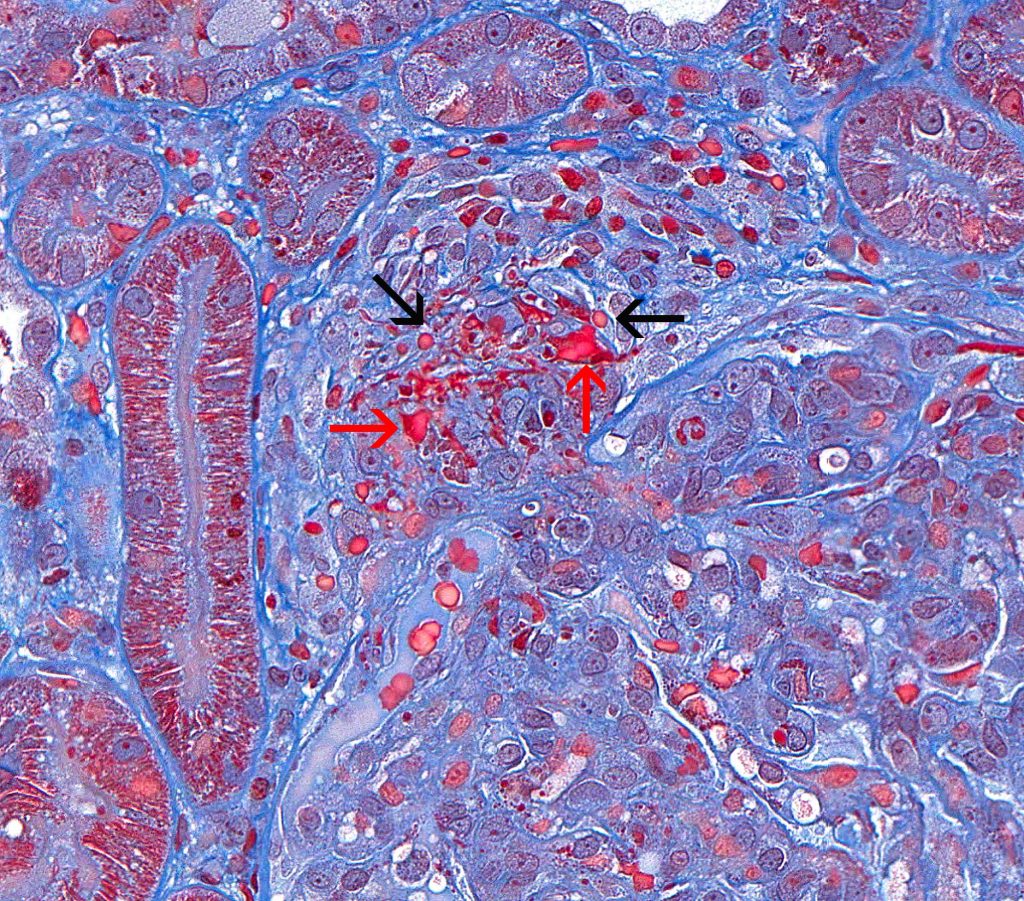 FIG.5G (MT): Higher magnification of FIG.5C. The fibrinoid necrosis can be seen as red material (red arrows), whereas there are also erythrocytes in the arteriolar wall (black arrows).
FIG.5G (MT): Higher magnification of FIG.5C. The fibrinoid necrosis can be seen as red material (red arrows), whereas there are also erythrocytes in the arteriolar wall (black arrows).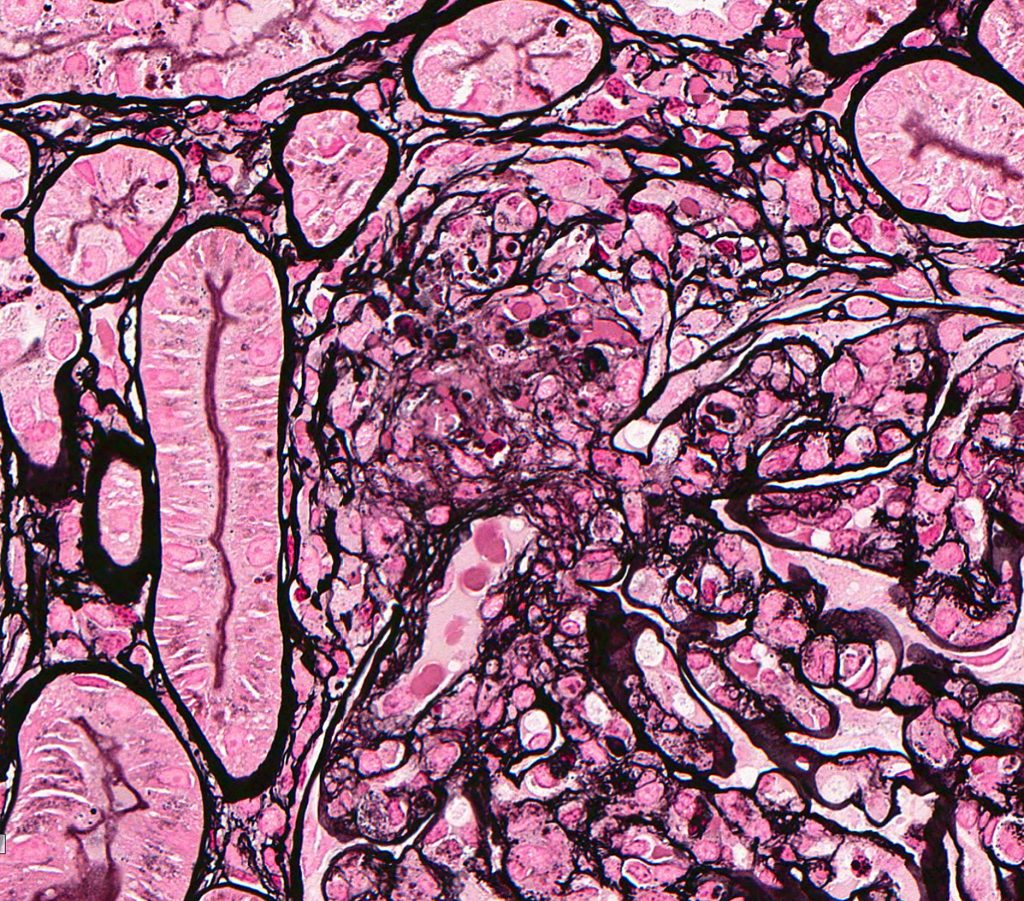 FIG.5H (JMS): Higher magnification of FIG.5D. The silver stain demonstrates the destruction of the basement membrane of the afferent arteriole.
FIG.5H (JMS): Higher magnification of FIG.5D. The silver stain demonstrates the destruction of the basement membrane of the afferent arteriole.![]()
FIG.5I (TEM): Endothelial cells are swollen, leading to loss of fenestrae and distortion of circulating red blood cells (RBC). There are small electron dense deposits in subendothelial and intramesangial regions.
FIG.5J (TEM): Colorized version of above TEM. Green: GBM; Yellow: Endothelial cell; Pink: Podocyte; Blue: Mesangium.
